SCIENCE丨扩展测序:完整生物系统中空间精确的原位转录组学
作者:马倩

本文通讯作者George M. Church是哈佛医学院遗传学教授,现代生物学领域最重要的意见领袖之一。研究方向集中在生物工程、基因组测序分析和合成生物学等方面。Adam H. Marblestone曾是George M. Church的博士研究生,目前主要研究生物纳米技术和合成生物学。Edward S. Boyden是麻省理工脑科学及认知科学方向的教授,在生物成像、生物分子工程、神经生物学以及组织工程等方向有着广泛的研究。
2014年,哈佛大学的George M. Church团队发表了荧光原位测序技术(FISSEQ)。首先,将RNA逆转录为cDNA,然后成环,扩增,交联形成细胞内3D原位RNA测序库。在FISSEQ中进行RNA的非靶向原位测序,将RNA放大成cDNA,在荧光显微镜上用标准的下一代测序化学方法测序。2015年,Edward Boyden开发了扩展显微镜技术(Expansion Microscopy,ExM)。Boyden运用的是丙烯酸类聚合物,原位形成凝胶,并用蛋白酶处理这种组织聚合物复合材料,以使其机械特性均匀化。最后注水就可以膨胀样品以用于成像。该方法线性放大比例约为4倍,适用于蛋白、RNA、代谢物等各种组织细胞内的生物分子。本文集合了FISSEQ和ExM这两种技术的优点,成功开发了Expansion sequencing(ExSeq)这一技术。ExM能够更好地分辨出密集的RNA转录本,用于原位杂交成像。
通过将 FISSEQ 扩增子的有效大小(200-400 nm)除以扩展因子,扩展样本有望使 FISSEQ 受益。这降低了扩增子的堆积密度,并有助于在多轮测序中对其进行跟踪。作者调整了 ExM 化学以在扩大的组织中启用 FISSEQ。特别是锚定(图 1Ai)、聚合(图 1Aii) 和扩展 (图 1Aiii) 步骤,将 RNA 分离以进行纳米级成像,导致整个可膨胀凝胶中存在带电的羧酸基团。通过将膨胀的样品重新嵌入不带电的凝胶中来稳定膨胀的样品,然后对样品进行化学处理以产生中性电荷环境。这将允许后续 FISSEQ 信号放大(图 1Aiv) 和读数 (图1B) 步骤进行。原位测序仅限于 5-30 个碱基的短读数。这种限制是由于成像过程中激光诱导的损伤以及给定周期的信号依赖于来自先前周期的信号(称为“定相”)。将这种短读数与基因组对齐是具有挑战性的。此外,短读长不容易捕获 mRNA 的复杂性,例如选择性剪接。因此,作者添加了后续一轮的非原位经典“下一代”测序(图 1Ci,)。
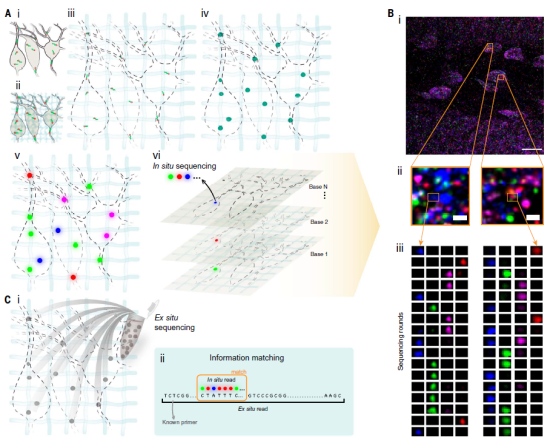
图1.非靶向扩展测序技术的概念和工作流程
为了验证 ExSeq,作者使用了以下小鼠标本:培养的海马神经元(图2A),一个 15 微米厚的海马切片(图 2C-D),以及 50 微米厚的海马切片 (图 2E-F)。为了提高 cDNA 环化的效率,将 cDNA 片段的大小限制在约 100 个碱基长。使用随机引物在与 50 微米厚 ExSeq 标本相邻的 50 微米厚海马切片上进行了 RNA 测序(RNAseq)(图 2E-F)。正如总 RNA 分析所预期的那样,在这两种情况下检测到的大部分 RNA 都是核糖体。作者观察到两种方法获得的 RNA 类型之间的总体一致性(图 2Gi); 其中 ExSeq 的编码 RNA 百分比略高(ExSeq 为 4-9%,RNAseq 为 2%)。在 FISSEQ 中,高丰度基因的代表性不足,例如参与翻译和剪接的基因。相比之下, 在ExSeq(28)中没有观察到这种检测偏差。ExSeq 和 RNAseq 之间的相关性随着成像的 ExSeq 体积而增加。通过采样模拟的更大体积产生更高的相关性(图 2Gvi)。在 10 个体积中,检测到 3,039 个基因,占通过 RNAseq 在样品中检测到的所有基因的约 16%,随着采样体积的增加而增加(图 2Gvii)。因此,ExSeq 能够以非靶向、高度多路复用的方式报告原位全基因组表达。
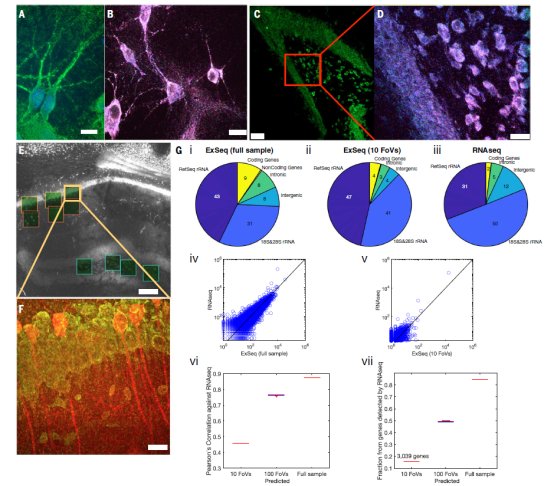
图2. 非靶向ExSeq在细胞和组织中进行原位测序
作者还使用非靶向扩展测序的方法对神经元转录本进行了亚细胞定位。利用 ExSeq 改进的空间分辨率来确定相对于抗体染色形态的 RNA。追踪了 13 个海马 CA1 锥体神经元。使用定制的 3D 查看器分析了已识别神经元内 RNA 的位置。神经元包含一个核与数千个突触。这就提出了一个问题,即 mRNA 的剪接,例如那些有助于突触功能的剪接,是否是沿着树突树以空间依赖的方式进行调节的。作者检查了对应于内含子区域的读数,并观察到,虽然 70% 的此类读数位于体细胞,但在树突下方可以找到包含 YFP 的树突投影中的内含子,这与之前的报告一致。例如,谷氨酸离子型受体红藻氨酸类型亚基 2 (Grik2) 编码参与兴奋性谷氨酸能神经传递的受体亚基,在文中的数据中出现在具有保留内含子的树突中。图 3Ai)。Grik1 亚基早先被鉴定为树突靶向的内含子保留序列。谷氨酸受体亚基 RNA 的树突剪接可能有助于调节兴奋性突触的状态或可塑性。事实上,树突中的剪接先前已在培养的神经元中表征。
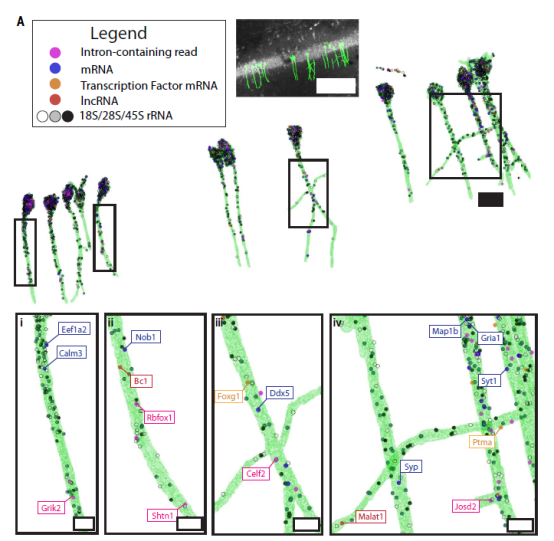
图3. 非靶向ExSeq在神经元树突中绘制RNA及其变体
非靶向测序能够在转录组范围内探索局部 RNA,包括稀有变异和功能未知的变异。然而,非靶向方法产生的读数的多样性导致检测到的分子的每个基因拷贝数较低,并且需要更多的生化和成像循环来区分读数。相比之下,靶向方法检测较小的预定义基因集,适用于绘制细胞类型和状态、原位空间关系以及亚细胞基因调控的可视化。作者绘制了小鼠初级视觉皮层的细胞类型,对其进行了基于单细胞 RNA 测序 (scRNA-Seq) 数据的细胞类型分类。设计了一组针对 42 个基因的探针,这些基因标记了关键的兴奋性和抑制性神经元类型。在 0.933 mm x 1.140 mm x 0.02 mm 的体积上对 Thy1-YFP 小鼠初级视觉皮层的冠状部分对这 42 个基因进行了靶向 ExSeq,测序了 265,347 个读数。 在小白蛋白阳性 (Pvalb + ) 中间神经元 (PV 中间神经元)、小白蛋白 (Pvalb)、囊泡抑制性氨基酸转运蛋白 (Slc32a1) 和谷氨酸脱羧酶 2 中(Gad2) 转录本共定位 (图 4C)。相比之下,与深层皮层兴奋性神经元(以及血管活性肠肽 (VIP + ) 中间神经元)相关的癫痫蛋白 6 同源物 (Sez6) 转录物不与 Pvalb、Slc32a1 和 Gad2 转录物共定位。图 4C)。使用k- means 聚类表达谱,并使用 t-Stochastic Neighbor Embedding (t-SNE) ( 61 ) (图 4D)。将获取的结果与之前对小鼠初级视觉皮层 scRNA-Seq 的研究进行了比较 (图 4E)。观察到视觉皮层中兴奋性神经元的典型逐层分层。9 个 ExSeq 兴奋性神经元簇对应于 7 个 scRNA-Seq 兴奋性神经元簇,但分组略有不同(图 4E)。可以发现抑制性神经元 ExSeq 集群与 scRNA-Seq 集群一对一匹配。例如,在皮层各层发现的两个生长抑素中间神经元簇,表达 Unc-13 同源物 C 的 SST 簇(簇 SST Unc13c)和表达软骨凝集素的 SST 簇(簇 SST Chodl),在两个数据集中都显着出现(图 4F-G)。一些抑制性神经元的 ExSeq 簇映射到多个 scRNA-Seq 簇上。例如, PV 和 GABAergic (-PV) 的两个 ExSeq 簇映射到多个 scRNA-Seq 簇上(图 4E)。
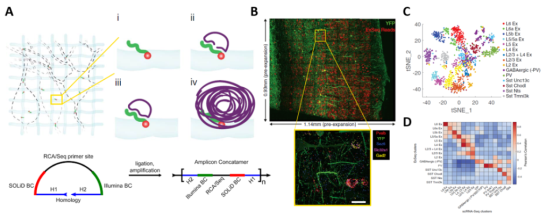
图4. 小鼠初级视觉皮层神经元转录本的靶向测序研究
接下来作者使用靶向 ExSeq 来探索小鼠海马神经元内的纳米级 RNA 区室化,其中树突状 RNA 与突触可塑性有关。追踪神经元中的 YFP 以识别树突和棘突,并针对之前在 CA1 神经元树突中发现的 34 个转录本进行测序。作者注意到在非靶向 ExSeq 海马数据中没有观察到刺,因为抗体染色是在测序后进行的,导致染色较弱,而这里的抗体染色是在扩增前进行的。使用 YFP 信号,对 CA1 锥体神经元和齿状回颗粒细胞进行了分割。在树突 (CA1, DG)、棘突 (CA1) 以及轴突 (CA1) 中发现了更小的转录本(图 5B)。在检查的 106,000 根棘中,作者在树突棘中发现了 730 个读数(每个棘有一个 RNA,除了一个有两个)。正如预期的那样,在 CA1 神经元中,突触后密度蛋白树突、突触可塑性相关基因 Camk2a 和突触后支架蛋白 SH3 和多个锚蛋白重复结构域 1 (Shank1) 等基因在树突中很突出。神经元钙传感器 Hpca 和突触谷氨酸受体 Gria1 是细胞体中最丰富的(图 5C)。在刺中, Shank1、腺苷酸环化酶 1 (Adcy1) 和驱动蛋白家族成员 5a (Kif5a) 是最丰富的转录本之一。作者发现细胞体、顶端树突、基底树突、顶端树突棘和基底树突棘中读数的分布在统计学上与其他各不相同,除了彼此没有区别的顶刺和基刺刺(图 5Ciii)。这表明在这些神经元中存在一组共同的脊柱 RNA 和脊柱 RNA 运输原则。
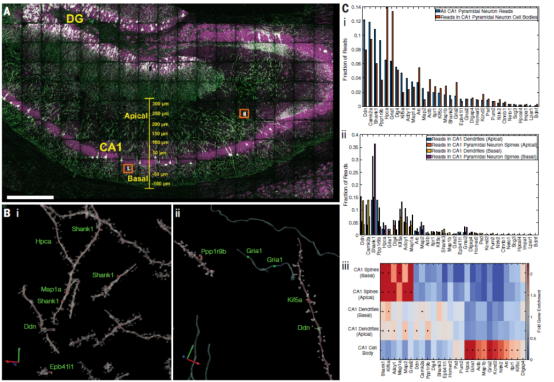
图5. 小鼠海马神经元树突和脊髓中纳米级转录组区隔化的靶向扩展测序特性
接下来探讨了 ExSeq 如何在癌症生物学和免疫学的背景下揭示基因表达的空间模式。一个关键问题是了解肿瘤微环境,包括免疫细胞的状态,如何控制肿瘤的生长、转移和治疗抵抗。对一名转移性乳腺癌浸润到肝脏的患者进行了核心活检,并对 297 个感兴趣的肿瘤相关基因进行了分析。解析了 115 万次读取,包括 2,395 个 DAPI 分段单元中的 771,904 次读取(图 6A)。ExSeq 的高 3D 空间分辨率允许在小于 1 微米的核结构内检测 516 个 RNA 读数,可能是核质桥,在组织中解析具有挑战性的结构。这些生物标志物包括在 B 细胞中发现的免疫球蛋白家族成员(IGHG1、IGHG4、IGKC),以及已知在转移性乳腺癌中表达的基因(孕酮受体,PGR;表皮生长因子受体,EGFR;醛脱氢酶 1 家族成员 A3, ALDH1A3 (图 6B)。肿瘤细胞和非肿瘤细胞高度混合(图 6C)。作者检查了细胞类型之间的空间共定位(图 6D)。不同的 B 细胞簇倾向于在空间中共定位,这与之前的观察结果一致。B 细胞簇表现出统计学显着性与所有其他细胞簇的共定位(图 6D) 除了一个表达基因标记物 PGR (Tumor PGR) 的肿瘤簇。这与 B 细胞直接与肿瘤细胞和巨噬细胞相互作用是一致的,这种相互作用有助于微环境中的体液反应。我们的分析还表明其他细胞类型的共定位,例如在成纤维细胞簇和巨噬细胞、T 细胞和肿瘤簇之间(图 6D)。因此,这种映射可能有助于阐明成纤维细胞在支持癌症部位白细胞聚集中的作用或成纤维细胞类型在癌症中的空间分布。作者最终分析了一种细胞类型是否可以根据与另一种细胞类型的物理接近程度来不同地表达基因。例如,一个单元格可能会根据物理接触或与另一个单元格的接近程度而改变状态。当 ALDH1A3 阳性肿瘤细胞靠近 HSPG2 阳性成纤维细胞时,低氧诱导因子 (HIF1A) 在 ALDH1A3 阳性肿瘤细胞中过表达 > 5 倍(图 6Eii)。基因 S100A8 是炎症过程和免疫反应的调节剂,可能是乳腺癌患者复发或进展的生物标志物,在 IGHG1 阳性 B 细胞中过度表达 4 倍,接近EGFR阳性肿瘤细胞(图 6Ei)。
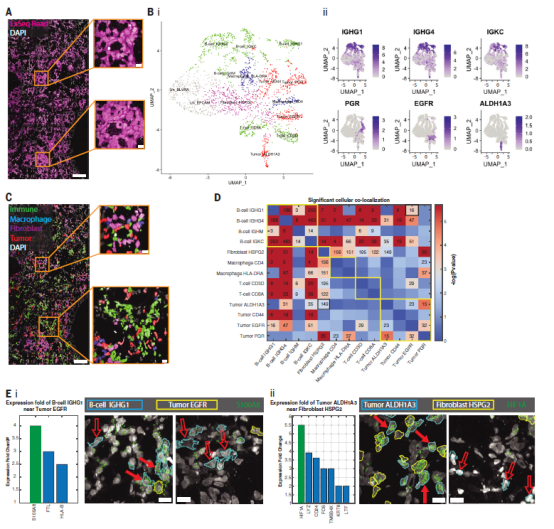
图6. 靶向ExSeq解决癌症中细胞类型和状态的映射
ExSeq 采用两种技术-扩增显微镜和原位测序-实现细胞和组织中 RNA 的空间精确、高度多重成像。非靶向和靶向形式的 ExSeq 有助于研究涉及亚细胞甚至纳米级 RNA 在完整细胞和组织环境中定位的科学问题(例如,如蛋白质的抗体染色或细胞核的 DAPI 染色所示)。它可以应用于多个器官系统和物种的标本,从小鼠大脑到人类癌症活检,以揭示细胞内部和细胞之间的空间关系。这些数据可能揭示细胞组织和功能的原理,并提供对复杂组织和多细胞系统中细胞如何相互作用或协调的潜在机制的见解。除了神经科学和癌症生物学,除了空间基因组学,ExSeq 可用于谱系和/或连接组索引 RNA 条形码的原位测序。
Alon S , Goodwin D R , Sinha A , et al. Expansion sequencing: Spatially precise in situ transcriptomics in intact biological systems. Science, 2021, 371(6528):eaax2656.

